A tutorial on Bayesian divergence-time estimation with SNP data
By Michael Matschiner with help from Marnus Stoltz.
Besides SVDQuartets (Chifman and Kubatko 2014), other methods for phylogenetic inference with the multi-species-coalescent model based on SNP data are implemented in SNAPP (SNP and AFLP Package for Phylogenetic analysis) (Bryant et al. 2012) and SNAPPER (Stoltz et al. 2021), two add-on packages for the program BEAST2. SNAPP is in principle similar to the approach of StarBeast3, only that each single SNP is considered as its own marker and gene trees are not separately inferred for each of these markers. Instead, SNAPP calculates the probability of the species tree without inferring gene trees, by mathematically integrating over all possible gene trees. This approach reduces the parameter space of the model tremendously and might thus be expected to also reduce the computational demand of the analysis. Unfortunately, however, the mathematical integration over all possible gene trees is computationally very demanding and SNAPP analyses are therefore only feasible for a relatively small number of individuals per species. This problem of SNAPP's high computational demand has recently been addressed with the release of SNAPPER, an approach that is similar to SNAPP except that a so-called diffusion model is used to calculate the likelihood of the species tree instead of mathematically integrating over all possible gene trees.
Another limitation of SNAPP has long been that the reported branch lengths were in coalescent units rather than in units of time, and that these could not easily be converted into time due to ascertainment bias in the SNP data. To address this, we tweaked the settings of SNAPP in our study Stange et al. (2018) so that they include a strict-clock model that can be time calibrated based on the fossil record or on information from other phylogenies.
- Outline
- Dataset
- Requirements
- Divergence-time estimation with SNAPP
- Divergence-time estimation with SNAPPER
- Interpretation of SNAPP results
- Comparison of results obtained with SNAPP and SNAPPER
In this tutorial I am going to present how the BEAST2 add-on packages SNAPP and SNAPPER can be used for divergence-time estimation with SNP data. The settings for time-calibrated inference with both approaches can not be specified through the program BEAUti, but instead the Ruby script snapp_prep.rb can be used to generate input files for time-calibrated analyses with SNAPP or SNAPPER. Run times will be compared between the two approaches, and differences between the inferred species trees and between these and the species tree generated in tutorial Bayesian Species-Tree Inference will be explored.
The SNP data used in this tutorial are the filtered dataset used for species-tree inference with SVDQuartets in tutorial Species-Tree Inference with SNP Data. You can find more information about the origin of this dataset in the Dataset section of this other tutorial. In brief, the dataset has been filtered to include only bi-allelic SNPs with a low proportion of missing data, for the 26 individuals from 13 cichlid species listed in the table below. Only SNPs mapping to chromosome 5 of the Nile tilapia genome assembly (Conte et al. 2017) are included in the dataset, and these have been thinned so that no pair of SNPs is closer to each other than 100 bp.
| Individual ID | Species ID | Species name | Tribe |
|---|---|---|---|
| IZA1 | astbur | Astatotilapia burtoni | Haplochromini |
| IZC5 | astbur | Astatotilapia burtoni | Haplochromini |
| AUE7 | altfas | Altolamprologus fasciatus | Lamprologini |
| AXD5 | altfas | Altolamprologus fasciatus | Lamprologini |
| JBD5 | telvit | Telmatochromis vittatus | Lamprologini |
| JBD6 | telvit | Telmatochromis vittatus | Lamprologini |
| JUH9 | neobri | Neolamprologus brichardi | Lamprologini |
| JUI1 | neobri | Neolamprologus brichardi | Lamprologini |
| KHA7 | neochi | Neolamprologus chitamwebwai | Lamprologini |
| KHA9 | neochi | Neolamprologus chitamwebwai | Lamprologini |
| IVE8 | neocra | Neolamprologus crassus | Lamprologini |
| IVF1 | neocra | Neolamprologus crassus | Lamprologini |
| JWH1 | neogra | Neolamprologus gracilis | Lamprologini |
| JWH2 | neogra | Neolamprologus gracilis | Lamprologini |
| JWG8 | neohel | Neolamprologus helianthus | Lamprologini |
| JWG9 | neohel | Neolamprologus helianthus | Lamprologini |
| JWH3 | neomar | Neolamprologus marunguensis | Lamprologini |
| JWH4 | neomar | Neolamprologus marunguensis | Lamprologini |
| JWH5 | neooli | Neolamprologus olivaceous | Lamprologini |
| JWH6 | neooli | Neolamprologus olivaceous | Lamprologini |
| ISA6 | neopul | Neolamprologus pulcher | Lamprologini |
| ISB3 | neopul | Neolamprologus pulcher | Lamprologini |
| ISA8 | neosav | Neolamprologus savoryi | Lamprologini |
| IYA4 | neosav | Neolamprologus savoryi | Lamprologini |
| KFD2 | neowal | Neolamprologus walteri | Lamprologini |
| KFD4 | neowal | Neolamprologus walteri | Lamprologini |
This tutorial requires BEAST2, Tracer, and FigTree to be installed. Details about the installation of these tools can be found in tutorial Bayesian Phylogenetic Inference.
The following tools are required additionally:
-
SNAPP: The SNAPP method (Bryant et al. 2012) implements a version of the multi-species coalescent model that mathematically integrates over all possible locus trees at biallelic loci, and is therefore particularly well suited for SNP data. SNAPP is an add-on package for BEAST2 and can be installed through the BEAST2 Package Manager. However, since BEAUti will not be used to prepare SNAPP input files, the SNAPP package does not need to be installed on local computers, but only on Saga. Use these commands to do so:
module purge module load Beast/2.7.0-GCC-11.3.0-CUDA-11.7.0 packagemanager -add SNAPP -
SNAPPER: The SNAPPER method (Stoltz et al. 2021) is similar to SNAPP in the sense that it uses biallelic loci as input and applies Bayesian MCMC to infer the species tree; however, the model used to calculate the tree likelihood is not the multi-species coalescent model but a Wright-Fisher diffusion model instead. Both of these models are approximations of one another and should produce similar results, particularly with larger population sizes. The difference between the two models is important when larger numbers of individuals are used per population, because the computational demand of SNAPP analyses increases rapidly with increasing numbers of individuals while that of SNAPPER remains roughly unchanged. Like SNAPP, SNAPPER is an add-on package for BEAST2 that can be installed through the BEAST2 Package Manager. The installation will also be required only on Saga and can be done with these commands:
module purge module load Beast/2.7.0-GCC-11.3.0-CUDA-11.7.0 packagemanager -add snapper
In this part of the tutorial, we are going to run a SNAPP analysis using an XML input file that is prepared with the Ruby script snapp_prep.rb (Stange et al. (2018)). The settings specified by this script allow time calibration of the species tree estimated by SNAPP through age constraints on one or more divergence events. Additionally, the population sizes of all species are linked to avoid unfeasible run times when more than a handful of species are analyzed.
Based on simulations, we tested the performance of SNAPP with a range of datasets in Stange et al. (2018). We found that the run time of SNAPP increases more or less linearly with the number of SNPs included in the dataset, but that the number of individuals used per species has an even stronger influence on run time. However, we also found that accurate and strongly supported species trees can be obtained with datasets containing only around 1,000 SNPs for just a single individual per species. Thus, before preparing the input file for SNAPP, we will first reduce the dataset that was already filtered in tutorial Species-Tree Inference with SNP Data. This further filtering will again be done with BCFtools.
-
If you no longer have file
NC_031969.f5.sub4.vcfin your current working directory on Saga (it was produced in tutorial Species-Tree Inference with SNP Data), either copy it from/cluster/projects/nn9458k/phylogenomics/week2/resor download it from GitHub, using one of the following two commands:cp /cluster/projects/nn9458k/phylogenomics/week2/res/NC_031969.f5.sub4.vcf .or
wget https://raw.githubusercontent.com/ForBioPhylogenomics/tutorials/main/week2_res/NC_031969.f5.sub4.vcf
As mentioned above and in tutorial Species-Tree Inference with SNP Data, the dataset in file NC_031969.f5.sub4.vcf contains SNP data for two individuals per species. To reduce the run time for SNAPP, we are going to produce another version of the same dataset that is reduced to only a single individual per species.
-
To exclude the individuals "IZA1", "AXD5", "JBD5", "JUI1", "KHA9", "IVF1", "JWH1", "JWG8", "JWH3", "JWH5", "ISA6", "IYA4", and "KFD4" (these were selected because they have more missing data than the other individuals of each species) from a new file named
NC_031969.f5.sub5.vcf, use the following commands:module purge module load BCFtools/1.12-GCC-10.2.0 srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty bcftools view -s ^IZA1,AXD5,JBD5,JUI1,KHA9,IVF1,JWH1,JWG8,JWH3,JWH5,ISA6,IYA4,KFD4 -o NC_031969.f5.sub5.vcf NC_031969.f5.sub4.vcfThe reduction of individuals might have led to some sites becoming monomorphic for either the reference or the alternate allele. Some sites might also have no data left for one or more of the species, given that these are now represented by only one instead of two individuals. These could now be excluded these sites again with BCFtools, for example with the
bcftools viewoption-e 'AC==0 || AC==AN || F_MISSING > 0.0'. However, this is not necessary because thesnapp_prep.rbscript, which will be used to write the XML file for SNAPP, will automatically exclude these sites anyway.Question 1: How many SNPs are left in file
NC_031969.f5.sub5.vcf? (see answer) -
Download the script
snapp_prep.rbfrom GitHub:wget https://raw.githubusercontent.com/mmatschiner/snapp_prep/master/snapp_prep.rb -
To see the options available for generating SNAPP input files with
snapp_prep.rb, have a look at the help text of the script by using the following commands:module load Ruby/2.7.2-GCCcore-10.2.0 ruby snapp_prep.rb -hYou'll see that
snapp_prep.rbcan write XML files for either SNAPP or SNAPPER (option-a), and that it accepts input either in Phylip (with option-p) or VCF (option-v) format. In addition, the script requires a table file (option-t) in which individuals are assigned to species, and a constraint file (option-c) in which age constraints for selected divergence events are specified. None of the other options are required, but these can be useful if you need to specify a starting tree (option-t) because SNAPP fails to find a suitable starting tree itself, if you want to increase or decrease the default length (500,000 iterations) of the MCMC chain (option-l), or if you want to limit the dataset to a certain number of SNPs to reduce the run time (option-m). -
Examples for the table and constraint files can be found on the GitHub repository for
snapp_prep.rb. Have a look at these example files; the example table file is namedexample.spc.txtand the example constraint file is namedexample.con.txt. -
To prepare the table file assigning individuals to species, open a new file named
individuals.txtwith a text editor available on Saga, and write the following text to this new file:species individual astbur IZC5 altfas AUE7 telvit JBD6 neobri JUH9 neochi KHA7 neocra IVE8 neogra JWH2 neohel JWG9 neomar JWH4 neooli JWH6 neopul ISB3 neosav ISA8 neowal KFD2 -
For the constraint file, we'll need to specify at least one age constraint for a divergence among the 13 cichlid species. For this, we can refer to the results of the analysis with the multi-species coalescent model in tutorial Bayesian Species-Tree Inference. In that tutorial, the divergence of Astatotilapia burtoni ("astbur") and the Neolamprologus species was estimated at 4.53 Ma with a 95% HPD interval from 3.21 to 6.09 Ma. We can approximate this mean and confidence interval with a lognormal distribution centered at 4.53 Ma that has a standard deviation (in real space) of 0.16. In addition, we could also constrain the first divergence among the species Neolamprologus brichardi ("neobri"), Neolamprologus gracilis ("neogra"), Neolamprologus marunguensis ("neomar"), and Neolamprologus olivaceus ("neooli") according to the results of this earlier analysis; however, it is likely that this divergence event would be more precisely estimated with the current SNP dataset. Thus, we are only going to constrain the age of a single node: the divergence of Astatotilapia burtoni ("astbur") from the species of the tribe Lamprologini.
To write this single age constraint to a constraint file, write the following text to a new file named
constraints.txt:lognormal(0,4.53,0.16) crown astbur,altfas,telvit,neobri,neochi,neocra,neogra,neohel,neomar,neooli,neopul,neosav,neowalNote that the above line contains three character strings that are delimited with white space (tabs or spaces). The first of the three character strings specifies that a lognormal distribution with an offset of 0, a mean of 7.02, and a standard deviation (in real space) of 0.13 should be used for the constraint. The second character string ("crown") specifies that the crown divergence of the clade should be constrained rather than the stem divergence (this would be the time at which the clade diverged from its sister group). Finally, the third character string simply lists all species included in the constrained clade, separated by commas. Because the constraint used here applies to the very first divergence of the phylogeny (the root), all species names are listed in the third character string.
Question 2: Can you think of a way how the exact same age calibration could be specified differently? (see answer)
-
With the VCF file
NC_031969.f5.sub5.vcf, the table file, and the constraints file ready, we can now prepare the input file for SNAPP with the scriptsnapp_prep.rb. To limit the dataset to 1,000 randomly selected SNPs and to set a chain length of 100,000 MCMC iterations, we'll use the options-m 1000and-l 100000, respectively. To specify that the XML file should be for SNAPP rather than SNAPPER, option-adoes not need to be specified because the SNAPP format is produced by default. Thus, use the following command to generate the XML input file for SNAPP:srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty ruby snapp_prep.rb -v NC_031969.f5.sub5.vcf -t individuals.txt -c constraints.txt -m 1000 -l 100000Question 3: How many sites useful for SNAPP did the dataset contain after we reduced it to a single individual per species? (see answer)
You may notice that the chain length of 100,000 MCMC iterations is extremely short compared to those used in the BEAST2 analyses of other tutorials. Using much shorter chain lengths with SNAPP than BEAST2 is quite common, given that the SNAPP model has far fewer model parameters than most models used in other BEAST2 analyses, and that each individual iteration is much slower with SNAPP due to the high computational demand of the integration over all possible gene trees at each SNP.
-
The script
snapp_prep.rbshould have written a file namedsnapp.xml. You could open that, for example withless -S snapp.xmland read some of the annotations if you'ld like to know more about the settings that we are about to use with SNAPP.
To "run SNAPP", we actually run BEAST2. The analysis can be sped up by using multiple CPUs, as SNAPP analyses are highly parallelizable. Thus, with e.g. four CPUs available that are all used for the SNAPP analysis, this analysis should take only about a fourth of the time that would be required with a single CPU. When running SNAPP analyses on a server such as Saga, one could even use tens of CPUs simultaneously, which would shorten SNAPP's run times tremendously. We will here use 16 CPUs on Saga to speed up the analysis.
-
Write a Slurm script named
run_snapp.slurmwith the following content to prepare the SNAPP analysis:#!/bin/bash # Job name: #SBATCH --job-name=snapp # # Wall clock limit: #SBATCH --time=2:00:00 # # Processor and memory usage: #SBATCH --ntasks=1 #SBATCH --cpus-per-task=16 #SBATCH --mem-per-cpu=1G # # Accounting: #SBATCH --account=nn9458k # # Output: #SBATCH --output=run_snapp.out # Set up job environment. set -o errexit # Exit the script on any error set -o nounset # Treat any unset variables as an error module --quiet purge # Reset the modules to the system default # Load the beast2 module. module load Beast/2.7.0-GCC-11.3.0-CUDA-11.7.0 # Run snapp. beast -threads 16 snapp.xml -
Submit the Slurm script with
sbatch:sbatch run_snapp.slurm -
Monitor the file
run_snapp.outby repeatedly opening it over the course of a few minutes (using, e.g.less run_snapp.outortail -n 1 run_snapp.out).Question 4: How long is SNAPP going to run to reach the MCMC length of 100,000 iterations that we specified when setting up the XML with
snapp_prep.rb? (see answer)
While the SNAPP analysis is running, we will in parallel set up an analysis with SNAPPER. This is easily done because SNAPPER analyses that are set up with snapp_prep.rb require the same input files as SNAPP analyses produced with the same script.
-
Re-run the script
snapp_prep.rbwith the same settings as before but now specifying-a SNAPPERto produce an XML file for SNAPPER instead of SNAPP:srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty ruby snapp_prep.rb -a SNAPPER -v NC_031969.f5.sub5.vcf -t individuals.txt -c constraints.txt -m 1000 -l 100000This should produce a new XML file named
snapper.xml -
Prepare a new Slurm script to launch the SNAPPER analysis, by copying the Slurm script for SNAPP:
cp run_snapp.slurm run_snapper.slurm -
Then, open the Slurm script
run_snapper.slurmin a text editor and change all occurrences of "snapp" to "snapper", so that the Slurm script has the following content:#!/bin/bash # Job name: #SBATCH --job-name=snapper # # Wall clock limit: #SBATCH --time=2:00:00 # # Processor and memory usage: #SBATCH --ntasks=1 #SBATCH --cpus-per-task=16 #SBATCH --mem-per-cpu=1G # # Accounting: #SBATCH --account=nn9458k # # Output: #SBATCH --output=run_snapper.out # Set up job environment. set -o errexit # Exit the script on any error set -o nounset # Treat any unset variables as an error module --quiet purge # Reset the modules to the system default # Load the beast2 module. module load Beast/2.7.0-GCC-11.3.0-CUDA-11.7.0 # Run snapper. beast -threads 16 snapper.xml(note that in this case, the command
cat run_snapp.slurm | sed "s/snapp/snapper/g" > run_snapper.slurmwould have produced the same result). -
Close and save the file and then submit the Slurm script with
sbatch:sbatch run_snapper.slurm
While the SNAPPER analysis (and probably also still the SNAPP analysis) are running, we can set up a second SNAPPER analysis to test how SNAPPER analyses are influenced by the use of additional individuals. As described above, the run time of SNAPPER should not increase substantially with additional individuals due to the implemented diffusion model. On the other hand, the precision of the results might be expected to increase. To run SNAPPER with additional samples, we'll use the original dataset in file NC_031969.f5.sub4.vcf, which contains SNP data for two individuals per species.
-
Because the set of individuals is now larger, we need to prepare a second table file assigning individuals to species. Open a new file named
individuals2.txtwith Emacs or another text editor:emacs individuals2.txt -
Write the following text to this new file:
species individual astbur IZA1 astbur IZC5 altfas AUE7 altfas AXD5 telvit JBD5 telvit JBD6 neobri JUH9 neobri JUI1 neochi KHA7 neochi KHA9 neocra IVE8 neocra IVF1 neogra JWH1 neogra JWH2 neohel JWG8 neohel JWG9 neomar JWH3 neomar JWH4 neooli JWH5 neooli JWH6 neopul ISA6 neopul ISB3 neosav ISA8 neosav IYA4 neowal KFD2 neowal KFD4 -
Save and close the file.
-
Write a new XML input file for SNAPPER, again using the script
snapp_prep.rb. This time, we'll use fileNC_031969.f5.sub4.vcfas input, and the new fileindividuals2.txtto assign individuals to species. To avoid overwriting the files for the running SNAPPER analysis, we'll have to additionally use the options-xand-o, setting the name of the XML file tosnapper2.xmland the prefix for the output files of SNAPPER tosnapper2:srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty ruby snapp_prep.rb -a SNAPPER -v NC_031969.f5.sub4.vcf -t individuals2.txt -c constraints.txt -m 1000 -l 100000 -x snapper2.xml -o snapper2Question 5: How many sites had to be excluded this time because they had only missing data in one or more species? (see answer)
-
Prepare a new Slurm script for the new SNAPPER analysis, by copying file
run_snapper.slurmand replacing all occurrences of "snapper" with "snapper2". The fastest way to do this is the following command:cat run_snapper.slurm | sed "s/snapper/snapper2/g" > run_snapper2.slurm -
Then, submit the new Slurm script with
sbatch:sbatch run_snapper2.slurm -
While the two SNAPPER analyses are running, monitor their output files
run_snapper.outandrun_snapper2.outby repeating the following commands a few times:tail run_snapper*.outAn estimate of the required run time per million iterations should be included in the output table after the first 6,200 iterations.
Question 6: How do the run times compare between the two analyses with SNAPPER? (see answer)
-
Once the SNAPP analysis has completed, download the file
snapp.logto your local computer withscpand open it in Tracer. The Tracer window should then display run statistics similar to those shown in the next screenshot.
You'll see that some low ESS values indicate that the MCMC chain has not yet reached stationarity and that the analysis should ideally have been performed with more MCMC iterations. For our interpretation here, however, we'll assume that the degree of stationarity is sufficient. What you also should notice is that the list of parameters on the left-hand side of the window is now much shorter than it usually is with results of BEAST2 analyses. In fact, only three parameters are shown: The speciation rate ("lambda"), the age of the root of the species tree ("treeHeightLogger"), and the substitution rate ("clockRate"). Note that the substitution rate is not comparable to a genome-wide rate due to ascertainment bias in the SNP dataset, even though SNAPP by default applies an ascertainment-bias correction (see Bryant et al. 2012 for details). However, one model parameter is not included in the log output yet, namely the population size. Recall that by using script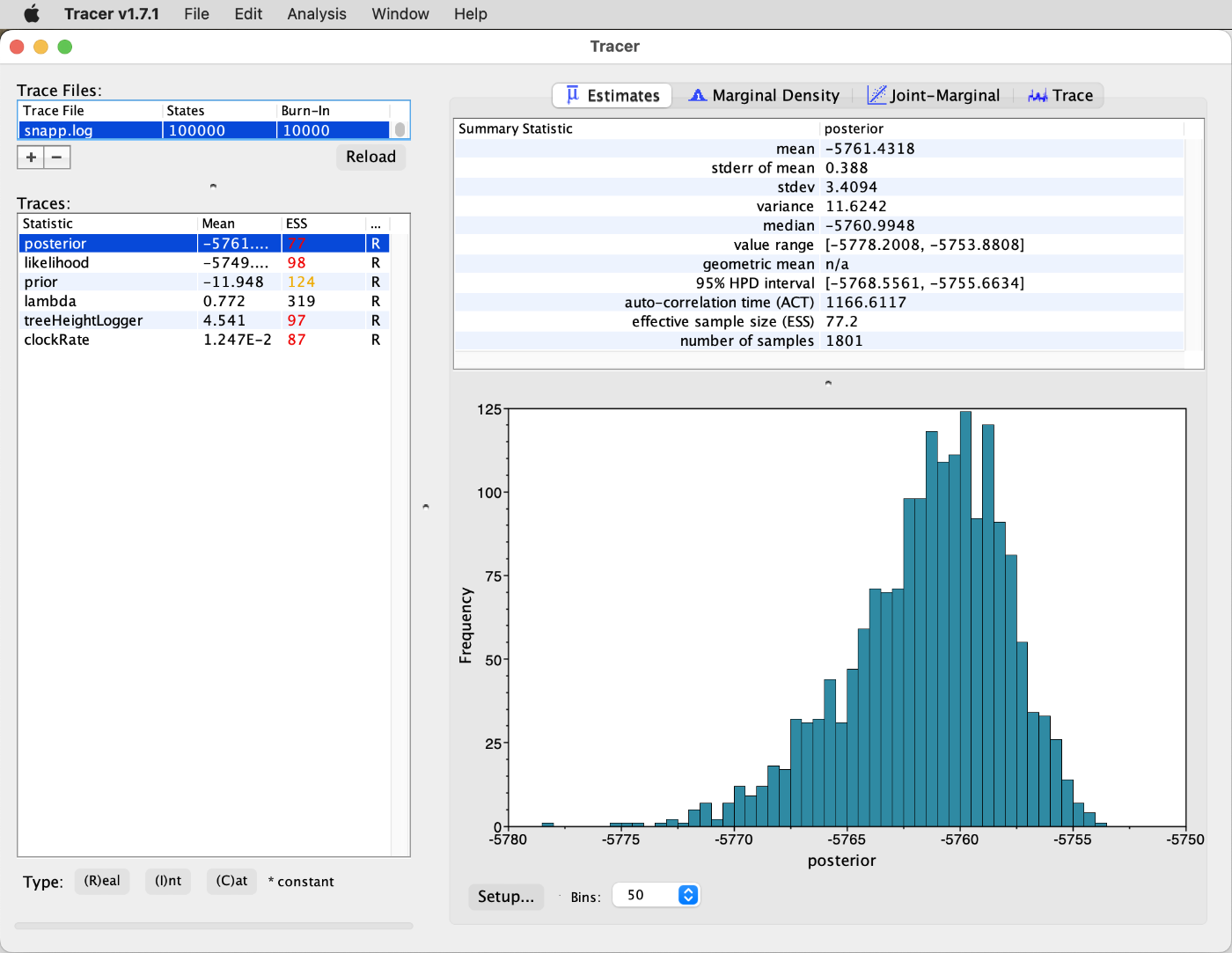
snapp_prep.rbto generate the XML file, we implemented a model in which the population sizes of all branches are set to be identical to each other. Thus, if the population sizes had been included in the log file, this file would contain a large number of columns with identical information. To avoid this, the output of the population sizes to the log file has been disabled bysnapp_prep.rb. However, the population size estimates are still available because they were instead written to the tree filesnapp.trees, and we can now add them to the log filesnapp.logusing the Ruby scriptadd_theta_to_log.rb. Thus, download the scriptadd_theta_to_log.rbfrom the GitHub repository forsnapp_prep.rbto your current directory on Saga:wget https://raw.githubusercontent.com/mmatschiner/snapp_prep/master/add_theta_to_log.rb -
Have a look at the available options for
add_theta_to_log.rb:ruby add_theta_to_log.rb -hYou'll see that this script requires four command-line arguments: The names of the log and tree input files, the name of an output file, as well as an estimate for the generation time. The latter is required to calculate an estimate of the effective population size, for which
add_theta_to_log.rbuses the equation Ne = Theta ÷ (4 × r ÷ ng), where r is the substitution-rate estimate (= the rate of the strict clock) and ng is the number of generations per time unit (more details are given in Stange et al. 2018). -
We'll here assume (as in other tutorials) that the generation time of cichlids is three years, and we'll name the output file
snapp_w_popsize.log. Thus use the following command to run scriptadd_theta_to_log.rb:srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty ruby add_theta_to_log.rb -l snapp.log -t snapp.trees -g 3 -o snapp_w_popsize.log -
Then, download file
snapp_w_popsize.logto your local computer and open it also in Tracer. You should see that "theta" and "population_size" have now been added to the list of parameters, as shown in the next screenshot.
-
Select both "clockRate" and "theta" from the list of parameters, click the tab for "Joint-Marginal" at the top right, and remove the tick at the bottom of the panel next to "Sample only". You should then see that both of these parameters are highly correlated, as shown in the next screenshot.
Note that both of these estimates should not be taken as being representative for the whole genome due to ascertainment bias. Because invariant sites are not included in a SNP dataset, this SNP dataset necessarily appears more variable than the whole-genome dataset from which it was derived, and thus the substitution rate of the SNP data appears to be higher than the rate would be if it was averaged over the entire genome. Nevertheless, our simulations in Stange et al. (2018) have shown that the population-size estimates of SNAPP are actually reliable.
-
Select "population_size" from the list of parameters and click on the "Estimates" tab to see summary information and a histogram for the population-size estimate, as shown in the next screenshot.

Question 7: How does this estimate of the population size of Lake Tanganyika cichlid fishes compare to that assumed in tutorial Bayesian Species-Tree Inference? (see answer)
-
Next, download the file
snapp.treesfrom Saga to your local computer and open it in the software Densitree from the BEAST2 package (it can be found in the same directory as the GUI versions of BEAST2 and BEAUti) to visualize the full set of posterior trees sampled by SNAPP, as shown in the next screenshot.
Unfortunately, the tip labels are misplaced due to a bug in the latest version of Densitree. Nevertheless, you should see that not all posterior trees share the same topology, indicating remaining uncertainty in the relationships of the 13 cichlid species. In particular, the relationships of Telmatochromis vittatus ("telvit"; the fifth species from the bottom) appear ambiguous, as this species is placed next to Neolamprologus walteri ("neowal") and Neolamprologus chitamwebwai ("neochi") in some of the posterior trees, but apparently closer to the remaining Neolamprologus species or ancestral to all of them in the other posterior trees. The relationships among the five species Neolamprologus brichardi ("neobri"), Neolamprologus olivaceous ("neooli"), Neolamprologus pulcher ("neopul"), Neolamprologus helianthus ("neohel"), and Neolamprologus gracilis ("neogra") also appear uncertain.
-
Also quantify the posterior probabilities of clades as node support in a maximum-clade-credibility tree using TreeAnnotator on Saga. Set the burnin percentage to 10, choose mean heights as node heights, select
snapp.treesas the input file, and name the output file "snapp.tre":module purge module load Beast/2.7.0-GCC-11.3.0-CUDA-11.7.0 srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty treeannotator -burnin 10 -heights mean snapp.trees snapp.tre -
Download file
snapp.trefrom Saga to your local computer withscpand open it in FigTree. -
Display the "posterior" node support values as node labels, as shown in the screenshot below.
The posterior probabilities for the different clades support the interpretation based on the Densitree plot made above: The position of Telmatochromis vittatus ("telvit") is uncertain, and so are the relationships within the clade comprising comprising Neolamprologus olivaceous ("neooli"), N. pulcher ("neopul"), N. gracilis ("neogra"), N. brichardi ("neobri"), and N. helianthus ("neohel").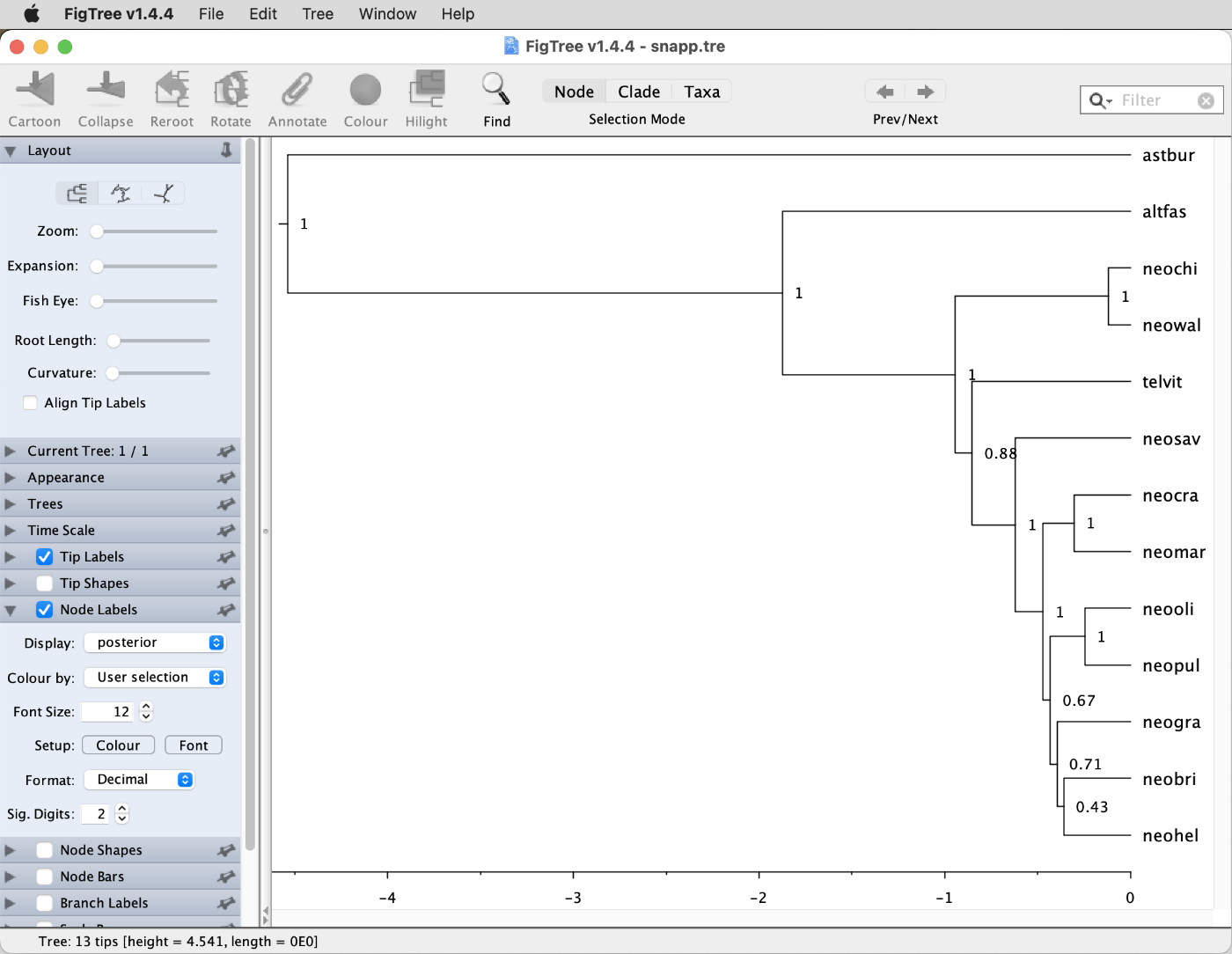
Question 8: Is the species-tree topology estimated with SNAPP concordant with that estimated with SVDQuartets in tutorial Species-Tree Inference with SNP Data? (see answer)
Question 9: How does the divergence-time estimate for Neolamprologus marunguensis ("neomar"), Neolamprologus gracilis ("neogra"), Neolamprologus brichardi ("neobri"), and Neolamprologus olivaceous ("neooli") compare to that obtained with StarBeast3 in tutorial Bayesian Species-Tree Inference? (see answer)






Despite the remaining uncertainty in the relationships among the Neolamprologus species, the SNAPP analysis has been valuable as it allowed us to estimate an average population size for species of the tribe Lamprologini, and improved the estimates of divergence times within the group.
Because the models used with SNAPP and SNAPPER were different, the likelihood, the prior probability, and the posterior probability are all not expected to be identical between the two analyses. However, the estimates for the speciation rate (lambda), the tree topology, and the divergence times should be similar with both tools. Additionally, we could expect that the estimates obtained by SNAPPER from the dataset with two individuals per species are more precise (have shorter confidence intervals) than those obtained with a single individual per species.
-
When the SNAPPER analyses have finished on Saga, generate maximum-clade-credibility trees with TreeAnnotator for both analyses:
module load Beast/2.7.0-GCC-11.3.0-CUDA-11.7.0 srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty treeannotator -burnin 10 -heights mean snapper.trees snapper.tre srun --ntasks=1 --mem-per-cpu=1G --time=00:01:00 --account=nn9458k --pty treeannotator -burnin 10 -heights mean snapper2.trees snapper2.tre -
Then, download the files
snapper.log,snapper2.log,snapper.tre, andsnapper2.treto your local computer. -
Open files
snapper.logandsnapper2.login Tracer together with filesnapp.log. Compare the ESS values for the parameters in the three files.Question 10: Does the MCMC chain from one of the three analyses appear more stationary than the others? (see answer)
-
Select all three trace files in the top left part of the Tracer window and the parameter "lambda" in the bottom left part. This should show a bar plot for the speciation-rate estimates from the three different analyses.

Question 11: Are the speciation-rate estimates comparable? And are those obtained with two individuals per species more precise than those with a single individual per species? (see answer)
-
Open the summary tree files
snapper.treandsnapper2.trein FigTree, together withsnapp.tre. Compare the age estimates between the three phylogenies. These might be as shown in the below three screenshots:
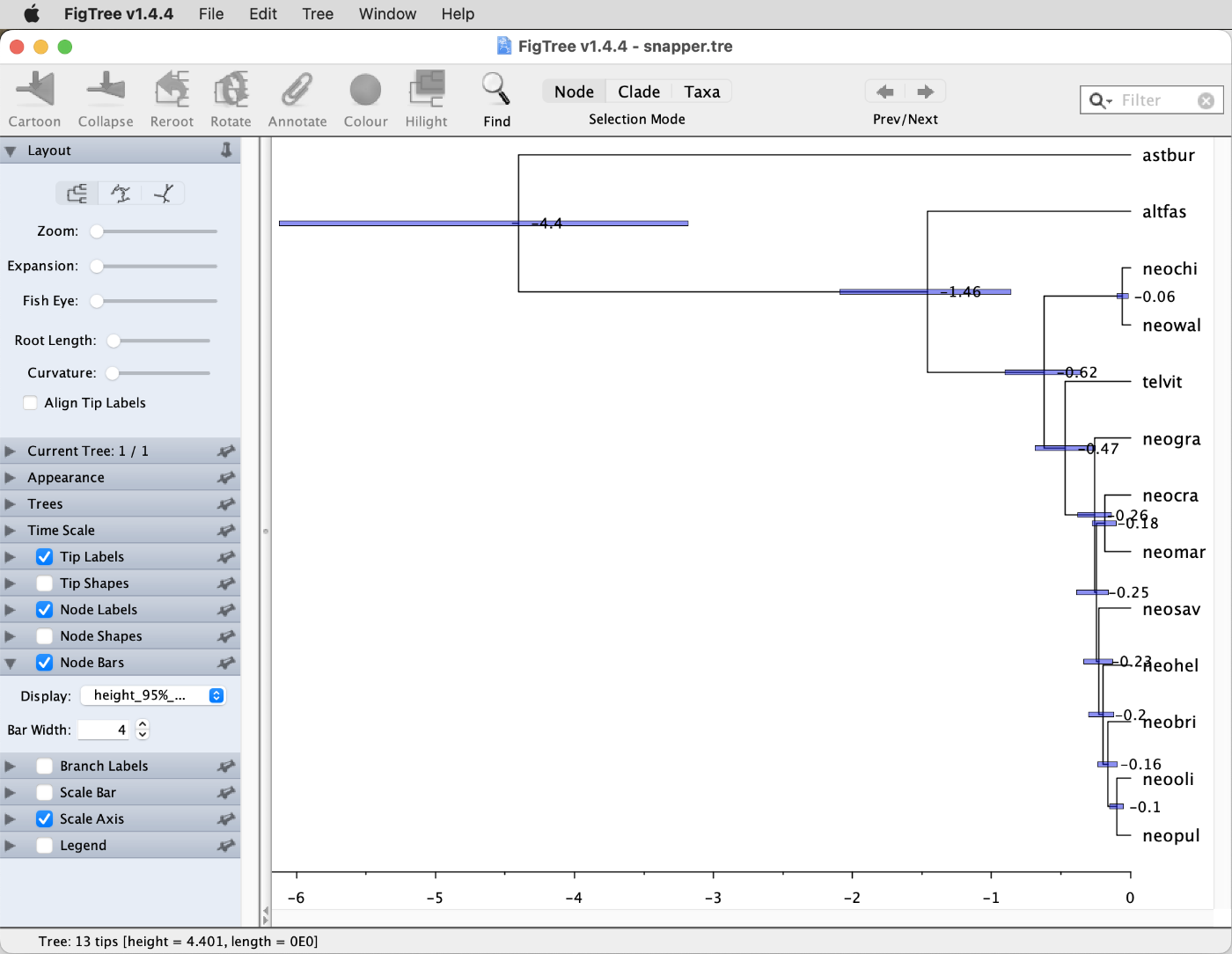

Question 12: Which differences do you notice in the age estimates? (see answer)
Question 13: Are there differences in the supported tree topology? (see answer)




-
Question 1: 65,325 SNPs should now remain in the filtered dataset in file
NC_031969.f5.sub5.vcf, which is more than sufficient for a phylogenetic analysis with SNAPP. To see the number of SNPs in this VCF file, you can use the the following command:bcftools view -H NC_031969.f5.sub5.vcf | wc -l
-
Question 2: Because the crown divergence of a clade is identical to the stem divergences of the two descending clades, we could also specify the same age constraint with the following line:
lognormal(0,7.02,0.13) stem altfas,telvit,neobri,neochi,neocra,neogra,neohel,neomar,neooli,neopul,neosav,neowalNote that "stem" is now specified as the second character string instead of "crown" and that "astbur" is now missing from the list of species in the third character string.
- Question 3: The screen output produced by
snapp_prep.rbshould report that 78 sites were half missing, 40,199 sites had only missing data for one or more species, and 6,931 sites were monomorphic out of the total of 65,325 sites. Thus, 65,325 - 78 - 40,199 - 6,931 = 18,117 sites could have been used by SNAPP. Out of these, the script sampled 1,000 sites because we had specified a maximum number of 1,000 sites with option-m.
- Question 4: You should be able to estimate the time required for the analysis by comparing the first column of the output in file
run_snapp.out, which reports the current MCMC iteration, with the time that the analysis has already been running. After a minute, the MCMC should have reached around iteration 3,000, which means that about half an hour will be required to complete the 100,000 iterations that we specified as the length of the MCMC. After about 6,000 iterations, the last column of the output in filerun_snapp.outshould also give an estimate of the run time required for 1 million iterations. Dividing this by ten should also result in a projected run time of around half an hour.
- Question 5: The screen output of
snapp_prep.rbshould report that 16,623 sites had to be exluded because they had only missing data in one or more species. This number is much lower than before when a single individual was used per species (40,199 sites). This shows that the addition of individuals can also help to increase the number of sites that can be used by SNAPP or SNAPPER.
- Question 6: The output of the two analyses should show that the SNAPPER analysis of file
snapper.xml(thus, with a single individual per species) requires around 9 hours per million iterations, and that the analysis of filesnapper2.xml(with two individuals per species) requires around 14 hours per million iterations. Thus, while the dataset has doubled, the run time has only increased by a factor of around 50%. Adding further individuals per species to the dataset would probably not increase the run time much further. For comparison, the SNAPP analysis required around 5 hours per million iterations and is thus faster than both SNAPPER analyses. But while we are not able to test this with our current dataset, it can be expected that with larger datasets including more individuals per species, the run times required for SNAPPER analyses should remain largely unchanged whereas those for SNAPP analyses grow more than linearly with the number of individuals (Stoltz et al. 2021).
- Question 7: Recall that in tutorial Bayesian Species-Tree Inference, the population-size parameter was fixed in the analysis with the multi-species coalescent model according to an estimate published by Meyer et al. (2017). In this study, Meyer et al. (2017) reported that "effective population sizes (Ne) estimated with the multispecies coalescent model ranged between 3.6 × 104 and 8.1 × 105, assuming a mean generation times of 3 years for cichlid fishes". Most of these population-size estimates in Meyer et al. (2017) were around 3.3 × 105, therefore this value was used in tutorial Bayesian Species-Tree Inference. In comparison to this population size assumed in the other tutorial, the current population-size estimate based on the SNAPP analysis, with a mean of 7.8 × 104 and a 95% HPD interval ranging from 5.3 × 104 to 1.1 × 105, is much smaller.
- Question 8: No, the topologies of the trees produced with SVDQuartets and SNAPP are not concordant. For example, SVDQuartets recovered a clade comprising Neolamprologus brichardi ("neobri"), N. olivaceus ("neooli"), and N. pulcher ("neopul"), even though Neolamprologus brichardi ("neobri") was found to cluster with N. helianthus ("neohel") in the SNAPP analysis. Note, however, that all conflicting nodes have low support in at least one of the two trees.
- Question 9: Recall that the divergence time of the four species Neolamprologus marunguensis ("neomar"), Neolamprologus gracilis ("neogra"), Neolamprologus brichardi ("neobri"), and Neolamprologus olivaceous ("neooli") was estimated around 0.82 Ma in the analysis with StarBeast3 in tutorial Bayesian Species-Tree Inference. In contrast, the first divergence of the clade comprising these four species was estimated with SNAPP at a more recent time, around 0.47 Ma, as shown in the screenshot below. The 95% HPD interval for this age estimate ranges from 0.31 to 0.65 Ma (select "height_95%_HPD" instead of "Node ages" from the drop-down menu to see these). In contrast, the 95% HPD interval for this divergence time in the StarBeast3 analysis was wider and ranged from 0.38 to 1.23 Ma, which thus includes almost the entire 95% HPD interval of the SNAPP analysis. This indicates that the two analyses do not disagree with each other regarding this divergence time but that the StarBeast3 analysis was not able to estimate it as precisely as the SNAPP analysis.


- Question 10: In my analyses, the ESS values were somewhat lower in the first SNAPPER analysis and far lower in the second, in which the burnin period had to be adjusted to around 30%.
- Question 11: At least in my analyses, the estimates for the speciation rate were all comparable, albeit slightly higher in the first SNAPPER analysis and, as expected, more precise in the SNAPPER analysis with two individuals per species.
- Question 12: In the results of my analyses, some nodes appeared to be quite a bit younger in the tree from the SNAPPER analysis with a single individual per species (
snapper.tre) than in the other two trees. The overall youngest divergence, however, was estimated in the SNAPPER analysis with two individuals per species, where the divergence of Neolamprologus chitamwebwai ("neochi") and N. walteri ("neowal") was estimated as young as 0.02 Ma or 20,000 years ago. The confidence interval for this divergence time was even extremely narrow, ranging from 0 to 0.04 Ma. To see whether the data really supports such a young age under the SNAPPER model, we would have to extend and replicate the anlaysis until it is fully stationary and converged.
- Question 13: A number of differences can be seen in the topologies of the three analyses; however, all of these affect nodes with low support values. The SNAPP analysis was the only one to recover a sister-group relationship between Neolamprologus helianthus ("neohel") and N. brichardi ("neobri"), but this relationship received a Bayesian posterior probability of only 0.43. Interestingly, the SNAPPER analysis with two individuals placed Telmatochromis vittatus ("telvit") next to the very young species pair Neolamprologus chitamwebwai ("neochi") and N. walteri ("neowal"), in contrast to the other two analyses.