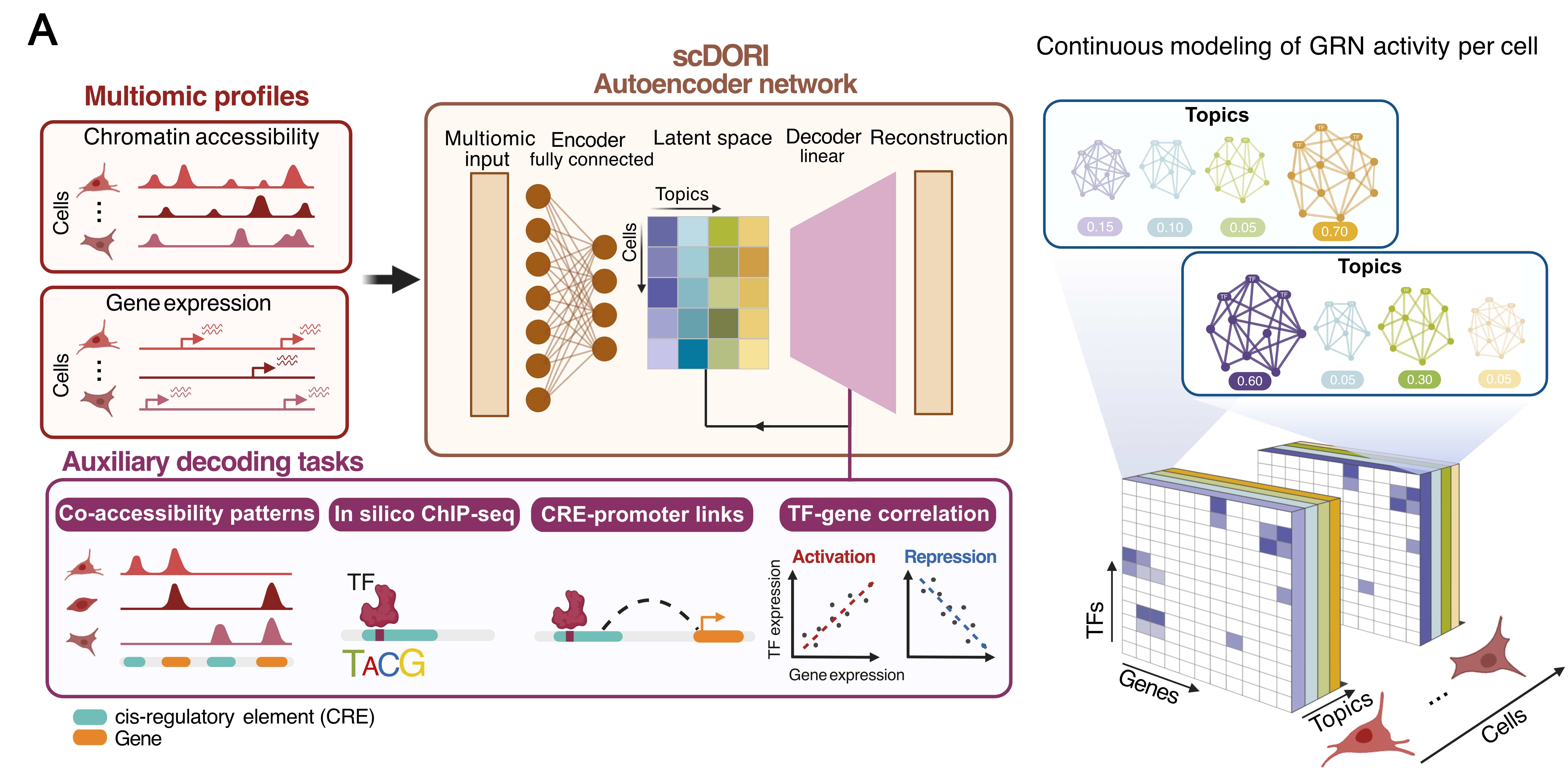
scDoRI is a deep learning model for single-cell multiome data (RNA + ATAC in same cell) that infers enhancer-mediated gene regulatory networks (eGRNs). By combining an encoder–decoder approach with mechanistic constraints (enhancer–gene links, TF binding logic), scDoRI learns topics that group co-accessible peaks, their cis-linked genes and upstream activator and repressor TFs – all while scaling to large datasets via mini-batches.
- 🔄 Unified approach: a single model for dimensionality reduction + eGRN inference
- 🧠 Learns topics that represent cell-state-specific regulatory programs
- 🧬Continuous eGRN modelling : each cell is a mixture of topics, allowing the study of changes in GRNs. No need for predefined clusters
- 🧰 Scalable to large datasets via mini-batch training
scDoRI expects single-cell multiome data with the following inputs:
RNA: an AnnData.h5adobject with cells × genes expression matrixATAC: an AnnData.h5adobject with cells × peaks accessibility matrix- Peaks must include genomic coordinates in
.var(columns:chr,start,end)
- Peaks must include genomic coordinates in
These datasets must be paired — i.e., RNA and ATAC should come from the same cells. Make sure that .X of anndata objects contain raw counts.
To install all dependencies for scDoRI, we recommend using Conda or Micromamba. The installation should take a couple of minutes.
git clone https://github.com/bioFAM/scDoRI.git
cd scDoRI
# create a new environment
conda create -n scdori python=3.12 -y
conda activate scdori
# Install BEDTools dependencies, e.g., via conda (if not installed on your system)
conda install --channel conda-forge --channel bioconda bedtools htslib -y
# Install the scDoRI package
pip install -e .Make sure the -e flag is used to install the package in editable mode.
The training process is GPU-accelerated and highly recommended to be run on a GPU-enabled machine. While preprocessing can run on CPU, training large datasets on CPU is not advised due to slow performance. If a GPU is available, the GPU-accelerated version will be installed automatically.
You’ll work through three notebooks - Preprocessing, Training and Downstream analysis. This will require two separate config files to set parameters for your dataset preprocessing and training.
src/scdori/pp/config.pyto specify the location of RNA and ATAC anndata .h5ad files, motif file, and set number of peaks/genes/TFs to train on.
docs/notebooks/preprocessing.ipynbThis step can take between a few hours to a day depending on the dataset size and number of features selected.
src/scdori/_core/config.pyfor scDoRI hyperparameters (number of topics, learning rate, epochs etc.) and specify path for preprocessed anndata objects and insilico-chipseq files
docs/notebooks/training.ipynbThis step again can take between a few hours to a day depending on the dataset size and number of features selected.
docs/notebooks/downstream.ipynbThe provided notebooks use the mouse gastrulation dataset from:
📄 Paper: Argelaguet et al., Bioarxiv 2022 📦 Download: Dropbox link
preprocessing_pipeline/config.py provides flexible options:
- You can set the number of peaks, genes, and TFs to use for model training
- 💡 Tip: Adjust based on your available GPU memory
- You can also force inclusion of specific genes or TFs, even if they aren’t highly variable
- Useful for focusing on known regulators/ genes of interest
📖 Full documentation and API reference is hosted at: https://scdori.readthedocs.io/en/latest/
Includes:
- API reference (docstrings)
- In-depth method overview
- Preprocessing + training guides
- (upcoming) Customization tips
If you use scDoRI in your work, please cite our preprint. Feel free to open an issue or get in touch at [email protected]