

- Song WZ, Zhang S, Thomas T* (2021) MarkerMAG: linking metagenome-assembled genomes (MAGs) with 16S rRNA marker genes using paired-end short reads (under review)
- Contact: Dr. Weizhi Song ([email protected]), Prof. Torsten Thomas ([email protected])
- Center for Marine Science & Innovation, University of New South Wales, Sydney, Australia
- 2022-03-12 - A demo dataset (together with command) has been prepared! You can use it to check if MarkerMAG is installed successfully on your system.
-
Workflow of MarkerMAG
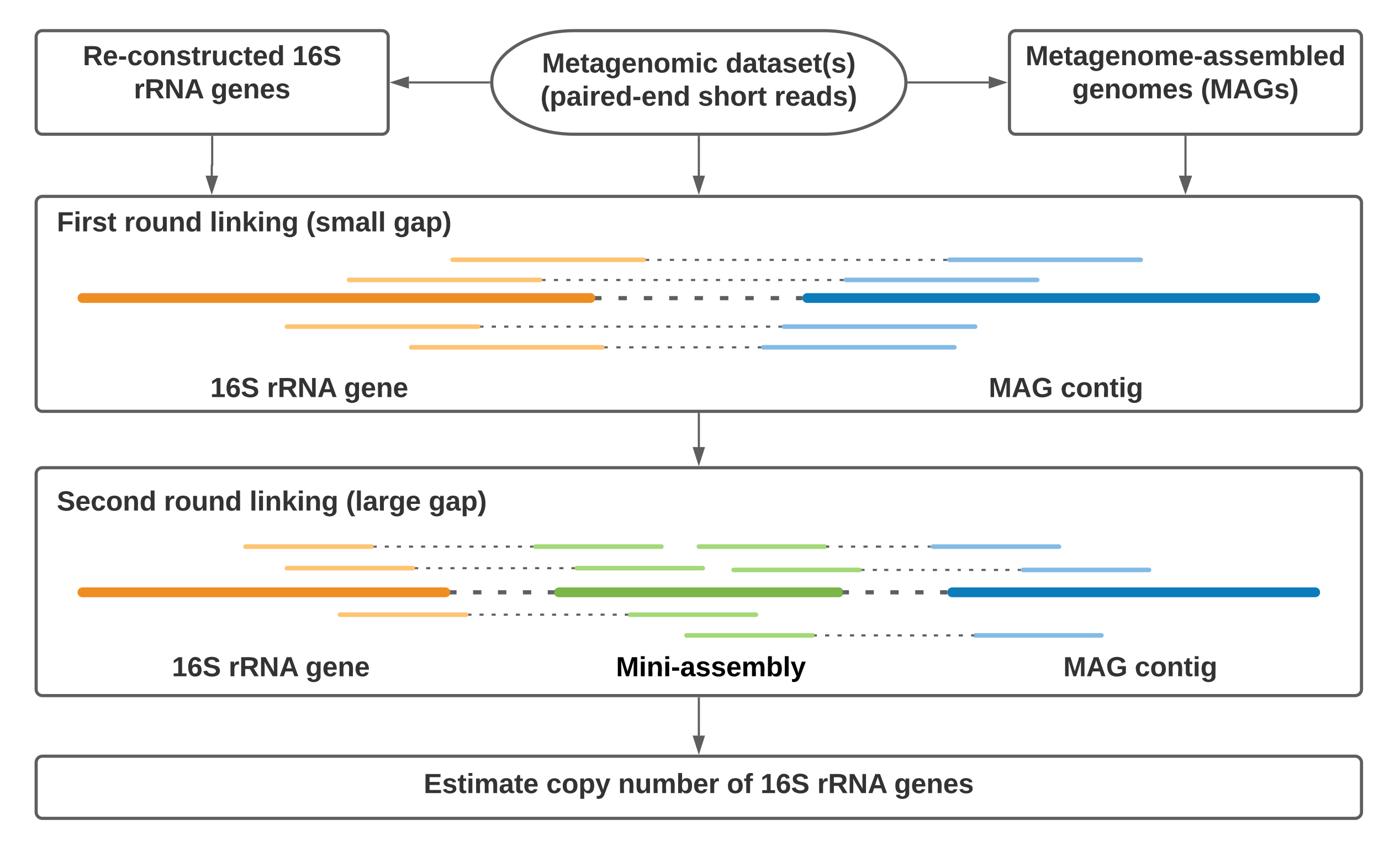
-
GC content bias
Read coverage of MAGs and their linked 16S rRNA genes might be biased by guanine-cytosine (GC) content [Reference]. Read coverage are weighted by GC content bias before estimating the copy number of 16S rRNA genes in MAGs. GC content bias is calculated as described here. An example of GC content bias from the MBARC-26 dataset that we used for benchmarking MarkerMAG is here.

-
Main module
link: linking MAGs with 16S rRNA marker genes
-
Supplementary modules
-
Dependencies for the
linkmodule: BLAST+, Barrnap, seqtk, Bowtie2, Samtools, HMMER, metaSPAdes, Usearch, as well as several Python packages, including Biopython, numpy, pandas, seaborn and plotly. -
Software dependencies need to be in your system path.
-
Dependencies for the supplementary modules can be found from the corresponding manual page.
-
MarkerMAG has been tested on Linux and MacOS, but NOT on Windows.
-
MarkerMAG is implemented in python3, it can be installed with pip3:
# install with pip3 install MarkerMAG # install a specific version of MarkerMAG (e.g. 1.1.15) pip3 install MarkerMAG==1.1.15 # upgrade with pip3 install --upgrade MarkerMAG -
⚠️ If you clone the repository directly off GitHub you might end up with a version that is still under development. -
Here are some example commands for UNSW Katana users.
-
MarkerMAG’s input consists of
- A set of user-provided MAGs
- A set of 16S rRNA gene sequences (either user-provided or generated with the
matam_16smodule) - The quality-filtered metagenomic reads used to generate the data above
-
⚠️ MarkerMAG is designed to work with paired short-read data (i.e. Illumina). It assumes the id of reads in pair in the format ofXXXX.1andXXXX.2. The only difference is the last character. You can rename your reads with MarkerMAG'srename_readsmodule (manual). -
Input reads to MarkerMAG need to be quality-filtered. If the input reads were provided in fastq format, MarkerMAG will first convert them into fasta format.
-
Although you can use your preferred tool to reconstruct 16S rRNA gene sequences from the metagenomic dataset, MarkerMAG does have a supplementary module (
matam_16s) to reconstruct 16S rRNA genes. Please refer to the manual here if you want to give it a go. -
Link 16S rRNA gene sequences with MAGs (demo dataset):
MarkerMAG link -p Demo -r1 demo_R1.fasta -r2 demo_R2.fasta -marker demo_16S.fasta -mag demo_MAGs -x fa -t 12
-
Summary of identified linkages at genome level:
Marker MAG Linkage Round matam_16S_7 MAG_6 181 Rd1 matam_16S_12 MAG_9 102 Rd1 matam_16S_6 MAG_59 55 Rd2 -
Summary of identified linkages at contig level:
Marker___MAG (linkages) Contig Round_1 Round_2 matam_16S_7___MAG_6(181) Contig_1799 176 0 matam_16S_7___MAG_6(181) Contig_1044 5 0 matam_16S_12___MAG_9(102) Contig_840 102 0 matam_16S_6___MAG_59(39) Contig_171 0 55 as well as its visualization:
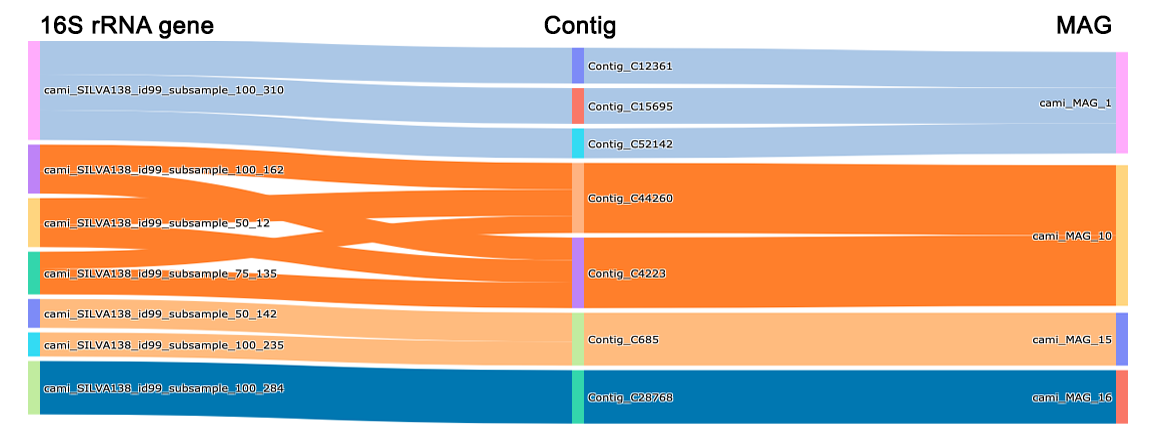
-
Copy number of linked 16S rRNA genes
MAG Copies MAG_9 1.97 MAG_59 3.41 -
Visualization of individual linkage
MarkerMAG supports the visualization of identified linkages (needs Tablet). Output files for visualization (example) can be found in the [Prefix]_linkage_visualization_rd1/2 folders. You can visualize how the linking reads are aligned to MAG contig and 16S rRNA gene by double-clicking the corresponding ".tablet" file. Fifty Ns are added between the linked MAG contig and 16S rRNA gene.
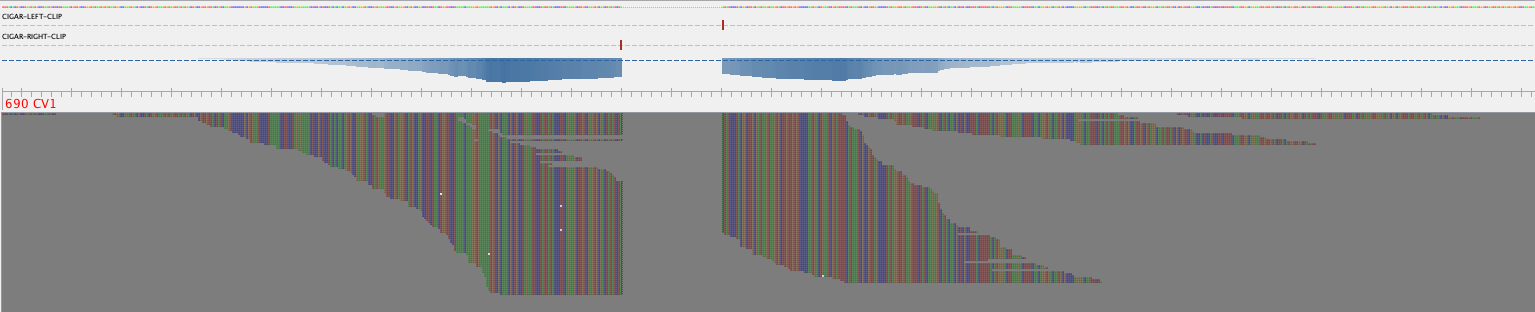
*If you saw error message from Tablet that says input files format can not be understood, please refer to here for a potential solution.

